




Unveiled: Earth’s Viral Diversity
NERSC Resources Help Researchers Identify 125,000 New Viral Sequences in Environmental Datasets
August 17, 2016
Contact: David Gilbert, degilbert@lbl.gov, 925-296-5643
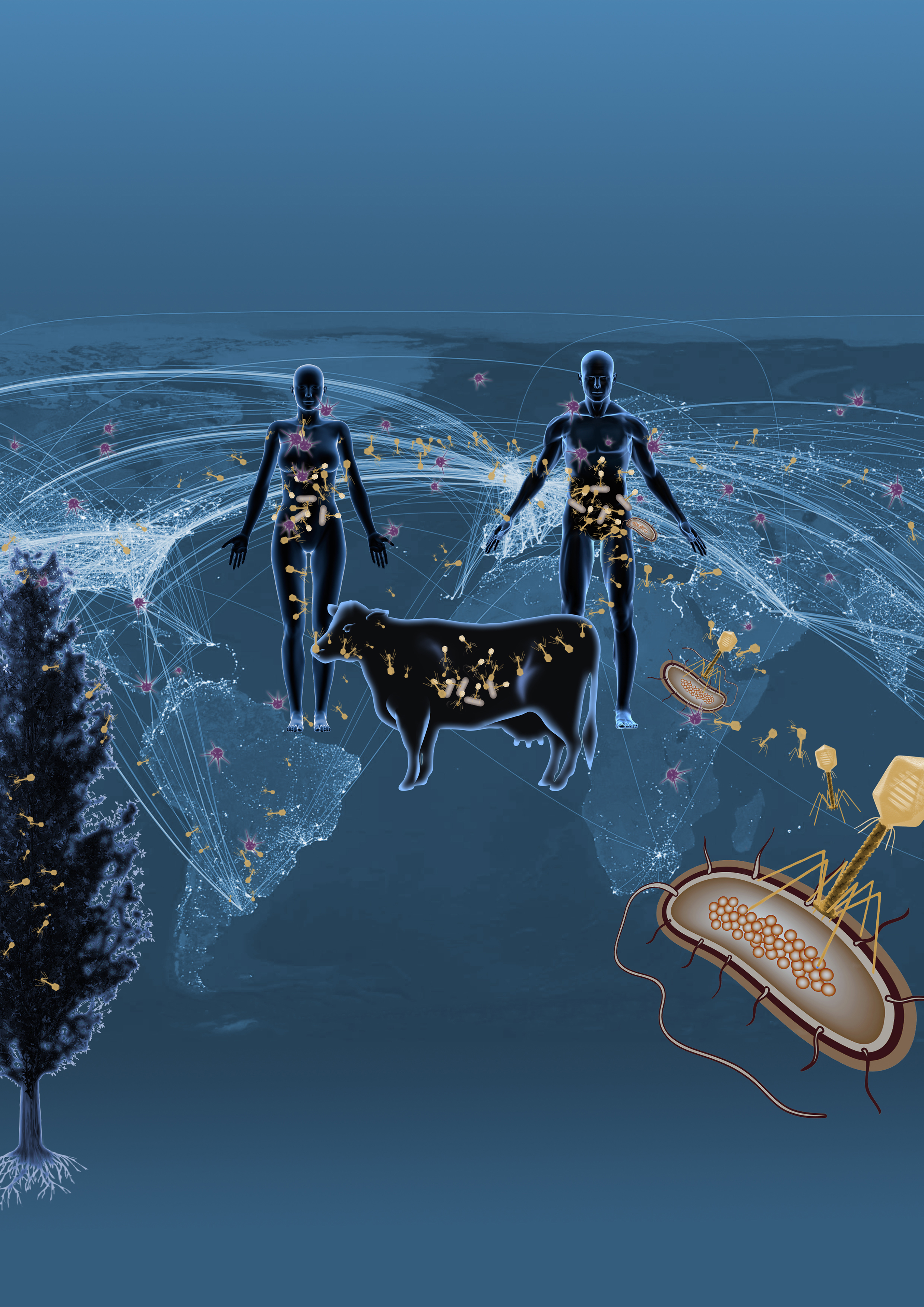
DOE JGI researchers utilized the largest collection of assembled metagenomic datasets from around the world to uncover over 125,000 partial and complete viral genomes, the majority of them infecting microbes. Graphic by Zosia Rostomian, Berkeley Lab
The number of microbes in, on, and around the planet – on the order of a nonillion, or 1030 – is estimated to outnumber the stars in the Milky Way. Microbes are known to play crucial roles in regulating carbon fixation, as well as maintaining global cycles involving nitrogen, sulfur, and phosphorus and other nutrients, but the majority of them remain uncultured and unknown. The U.S. Department of Energy is targeting this “microbial dark matter” to better understand the planet’s microbial diversity and glean from nature lessons that can be applied toward energy and environmental challenges.
Plumbing the Earth’s microbial diversity, though, requires learning more about the poorly-studied relationships between microbes and the viruses that infect them, viruses that impact the microbes’ abilities to regulate global cycles. Although the number of viruses is estimated to be at least two orders of magnitude more than the microbial cells on the planet, there are currently less than 2,200 sequenced DNA virus genomes, compared to the approximately 50,000 bacterial genomes, in sequence databases. In a study published online August 17, 2016 in Nature, researchers at the U.S. Department of Energy Joint Genome Institute (DOE JGI), a DOE Office of Science User Facility, utilized the largest collection of assembled metagenomic datasets from around the world to uncover over 125,000 partial and complete viral genomes, the majority of them infecting microbes. This single effort, which utilized NERSC's Genepool system – a cluster dedicated to JGI's computing needs – increases the number of known viral genes by a factor of 16 and provides researchers with a unique resource of viral sequence information.
“It is the first time that someone has looked systematically across all habitats and across such a large compendium of data,” said study senior author and DOE JGI Prokaryote Super Program head Nikos Kyrpides. “A key to uncover all these novel viruses was the sensitive computational approach we have developed along this work.”
Uncovering Novel Viruses
This approach, explained first author and postdoctoral fellow David Paez-Espino, involved using non-targeted metagenomics, referencing both isolate viruses and manually curated viral protein models, and what he described as “the largest and most diverse dataset to date.” The team analyzed over 5 trillion bases (Terabases or Tb) of sequence available in the DOE JGI’s Integrated Microbial Genomes with Microbiome Samples (IMG/M) system collected from 3,042 samples around the world from 10 different habitat types. Their efforts to sift through the veritable haystack of datasets yielded over 125,000 viral sequences containing 2.79 million proteins.
"Our estimate is that it took us about 10 million core-hours for all the computations against 4,000 metagenomes and around 50,000 isolate genomes," Kyrpides said. "It would have been extremely difficult, if not impossible, to do this work without NERSC's support."
The team matched viral sequences against multiple samples in multiple habitats. For example, one viral group they identified was found in 95 percent of all samples in the ocean’s twilight zone – a region located between 200 and 1,000 meters below the ocean surface where insufficient sunlight penetrates for microorganisms to perform photosynthesis.
By analyzing a CRISPR-Cas system – an immune mechanism in bacteria that confers resistance to foreign genetic elements by incorporating short sequences from infecting viruses and phages – the team was able to generate a database of 3.5 million spacer sequences in IMG. These spacers, fragments of phage genetic sequences retained by the host, can then be used to explore viral and phage metagenomes for where the fragments may have originally come from. Also, using mainly this approach, the team computationally identified the host for nearly 10,000 viruses. “The majority of these connections were previously unknown, and include the identification of organisms serving as viral hosts from 16 prokaryotic phyla for which no viruses have previously been identified,” they reported.
Beacons for CRISPR-Cas proteins
Jan-Fang Cheng, head of the DOE JGI’s Functional Genomics group, said the work being done by Kyrpides’ group in identifying new viral sequences will help the Synthetic Biology group develop novel promoters that can work in many bacterial hosts. “We are constantly searching for regulatory DNA parts that will work across many different phyla, and that would allow us to build genes and pathways that can express in many different hosts.”
Cheng also anticipated that the expanded viral sequence space generated by Kyrpides’ team will allow researchers to look for other genetic sequences known as proto-spacer adjacent motifs (PAMs). These sequences lie next to spacer sequencers in phages and are used as beacons by CRISPR-Cas proteins, triggering actions such as editing or regulating a gene. “People are looking for new PAM sequences and new Cas9s, and with this new information, if you can map the spacer sequence back to the same phage and align them and see what’s in common in neighboring sequences, then you could ID new PAM sequences.”
“We believe that the finding of many large phages including the longest phage genome reported thus far points to the limitations of conventional virome enrichment and sequencing strategies which may bias the studies against the highly novel viruses with unusual properties”, said Natalia Ivanova, group lead in the Super Program and co-author of this study.
“One of the most important aspects of this study is that we did not focus on a single habitat type. Instead, we explored the global virome and examined the flow of viruses across all ecosystems," said Kyrpides. We have increased the number of viral sequences by 50x, and 99 percent of the virus families identified are not closely related to any previously sequenced virus. This provides an enormous amount of new data that would be studied in more detail in the years to come. We have more than doubled the number of microbial phyla that serve as hosts to viruses, and have created the first global viral distribution map. The amount of analysis and discoveries that we anticipate will follow this dataset cannot be overstated.”
The work used resources at the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility at Lawrence Berkeley National Laboratory.
***
The U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility at Lawrence Berkeley National Laboratory, is committed to advancing genomics in support of DOE missions related to clean energy generation and environmental characterization and cleanup. DOE JGI, headquartered in Walnut Creek, Calif., provides integrated high-throughput sequencing and computational analysis that enable systems-based scientific approaches to these challenges. Follow @doe_jgi on Twitter.
About NERSC and Berkeley Lab
The National Energy Research Scientific Computing Center (NERSC) is a U.S. Department of Energy Office of Science User Facility that serves as the primary high performance computing center for scientific research sponsored by the Office of Science. Located at Lawrence Berkeley National Laboratory, NERSC serves almost 10,000 scientists at national laboratories and universities researching a wide range of problems in climate, fusion energy, materials science, physics, chemistry, computational biology, and other disciplines. Berkeley Lab is a DOE national laboratory located in Berkeley, California. It conducts unclassified scientific research and is managed by the University of California for the U.S. Department of Energy. »Learn more about computing sciences at Berkeley Lab.