




Mathematical Models Shed New Light on Cancer Mutations
Calculations Run at NERSC Pinpoint Rare Mutants More Quickly
November 3, 2014
Contact: David Cameron, 617.432.0441, david_cameron@hms.harvard.edu
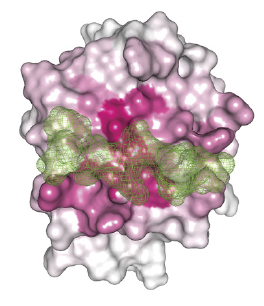
Heat map of the average magnitude of interaction energies projected onto a structural representation of SH2 domains (white) in complex with phosphopeptide (green). SH2 (Src Homology 2) is a protein domain found in many intracellular signal-transducing proteins. Phosphopeptides are modified self antigens that can induce an immune response. Image: M. AlQuraishi, et al, Harvard Medical School
A team of researchers from Harvard Medical School, using computing resources at the U.S. Department of Energy’s National Energy Research Scientific Computing Center (NERSC), have demonstrated a mathematical toolkit that can turn cancer-mutation data into multidimensional models to show how specific mutations alter the social networks of proteins in cells.
From this they can deduce which mutations among the myriad mutations present in cancer cells might actually play a role in driving disease. Their latest findings—among the first to be produced from the new Laboratory of Systems Pharmacology (LSP) at Harvard—were published November 2 in Nature Genetics.
At the core of this approach is an algorithm based on statistical mechanics, a branch of theoretical physics that describes large phenomena by predicting the macroscopic properties of its microscopic components. Lead author Mohammed AlQuraishi, an independent HMS Systems Biology fellow associated with the LSP, previously published on this method in 2012 with Harley McAdams of Stanford University (see “New mathematical method reveals where genes switch on or off”). In that study, which also utilized NERSC resources, they modeled atomic-level details of protein-DNA interactions with unprecedented accuracy; this new research is a follow-on that leveraged improvements in the mathematical method, AlQuraishi said.
“Here we have found that a fundamental concept in statistical mechanics, which many of us learned as undergraduates in theoretical physics courses and then largely forgot because it didn’t apply to our everyday lives as biologists, can be relevant to one of the most difficult problems in cancer genetics,” said Peter Sorger, professor of systems pharmacology at Harvard Medical and senior author on the paper.
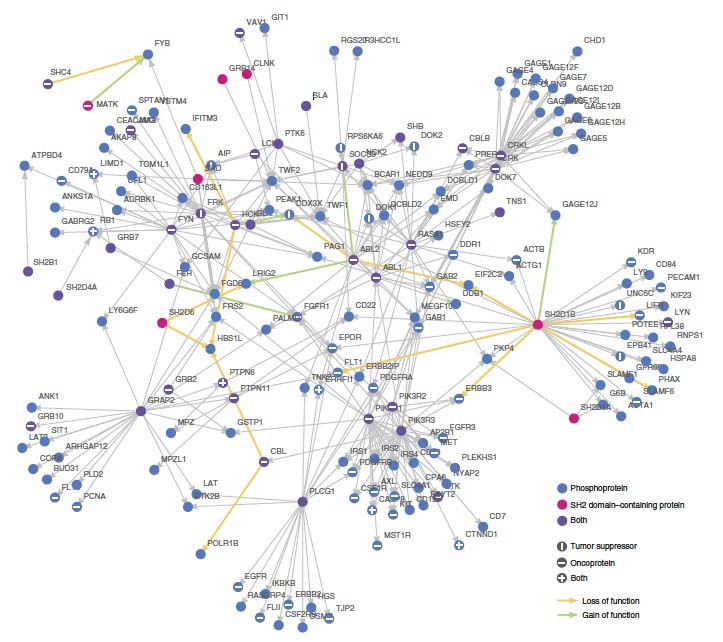
Kidney tumor network, generated using statistical mechanics, the Mathematica software package and data from the Cancer Genome Atlas. Image: M. AlQuraishi, et al, Harvard Medical School
Statistical Mechanics, Detailed Schematics
Many of the most widely studied cancer genes, such as P53 and Ras, were discovered after decades of work by many groups. But today, in the era of high throughput genomics, we have thousands of times more data from thousands of samples. As a result, the sheer volume of catalogued cancer mutations is vast.
But not all mutations actually influence tumor behavior. Many appear to be along for the ride, so to speak, and are as a result called “passenger mutations.” To separate the drivers from the passengers, researchers typically use a kind of “polling” strategy in which they identify the most common mutations, reasoning that those are the significant ones. Only the most promising candidates are then subjected to the detailed and painstaking analysis that has been applied to P53 and Ras.
AlQuraishi reasoned that biologists were in dire need of much more biophysically rigorous tools for scouring this data. With a background in genetics, statistics and physics, he realized that biologists can exploit the statistical power from live data sets and marry it to theoretical physics. “It’s the way that Silver and Feynman together would do it,” he joked.
Statistical mechanics is a precise physical description of how collections of individual molecules give rise to the macroscopic properties we perceive, such as temperature and pressure. AlQuraishi used its core principles as the basis for a platform that would analyze information housed in the National Cancer Institute’s Cancer Genome Atlas. As a result, he was able to generate detailed schematics of how certain mutations altered the vast, complex cellular world of protein social networks—networks that largely determine a cell’s health, or lack thereof. In doing so, he stumbled upon a few unexpected findings. For example, while many cancer mutations are common, many more are rare—some so rare that they only occur in a handful of patients. AlQuraishi found that common and rare mutations are equally likely to affect the protein network.
“Both kinds of mutations are equally strong,” he said. “In both cases, about 1 percent of the common and 1 percent of the rare mutations alter the tumor networks we studied. But rare mutations are being largely ignored. We need to start paying attention to them.”
Reproducing Results
The researchers also found that mutations are not really the blunt force that they expected. Rather than knocking out an entire branch of a network—such as a neighborhood power outage—or inserting an entirely new character (a protein), mutations cause a subtle, almost surgically precise, altering of the communication pathway.
“From the perspective of the mutation, it is hard to be so precise,” said AlQuraishi. “But cancer can’t be too disruptive, or else it might die. It needs to fly under the radar. This subtle altering of networks achieves that objective. Drug companies can exploit this and possibly develop more targeted therapies.”
A final area that these findings address is the problem of reproducing published results in the scientific literature. Here, however, the researchers are able to use fundamental physical principles to process datasets from different laboratories (including their own) in a way that removes the false positives and enriches for the true positives. The model is therefore more accurate and reproducible than any single data set.
“We can clean up the experiments by only using data that both the model and experiments agree on,” said AlQuraishi.
Exploring the large space of biophysical models he and his colleagues considered for this study required access to supercomputers with substantial amounts of memory, exceeding several hundreds of gigabytes on a single node, he emphasized.
“Without the systems available at NERSC, we would not have been able to perform these calculations in memory, if at all," he said. "This capability allowed us to explore much richer and more complex models more quickly, and ultimately led to the high performance of the algorithm.”
This article was adapted from materials provided by Harvard Medical School.
About NERSC and Berkeley Lab
The National Energy Research Scientific Computing Center (NERSC) is a U.S. Department of Energy Office of Science User Facility that serves as the primary high performance computing center for scientific research sponsored by the Office of Science. Located at Lawrence Berkeley National Laboratory, NERSC serves almost 10,000 scientists at national laboratories and universities researching a wide range of problems in climate, fusion energy, materials science, physics, chemistry, computational biology, and other disciplines. Berkeley Lab is a DOE national laboratory located in Berkeley, California. It conducts unclassified scientific research and is managed by the University of California for the U.S. Department of Energy. »Learn more about computing sciences at Berkeley Lab.