




Efficient Graph Analytics for Genomics
DNA sequencing is on an exponential trajectory, with the number of human genomes alone projected to double every 7 months. Current protocols do not attempt de novo assembly because it is too computationally burdensome, and instead rely on the simpler strategy of aligning short reads to a human “reference” genome. These approaches are useful for identifying local differences of one or a few letters between a target genome and the reference, but are inadequate for discovering larger differences, which may include the rearrangement, deletion or duplication of several genes, or for accessing regions of the genome that are highly variable between individuals.
De novo assemblers reconstruct a genome or (in the case of metagenomics) an unknown set of genomes from a collection of overlapping and erroneous DNA segments, relying on constructing and traversing a graph (de Bruijn, overlap, or string). De novo assembly is among the most computationally demanding tasks in bioinformatics.
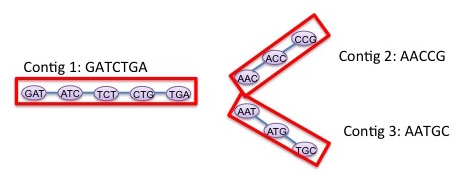
Partly funded via MANTISSA, we have developed parallel algorithms for de Bruijn graph construction and traversal using 2nd generation short-read sequence data. Our algorithms scale to an unprecedented O(100K) cores for large enough datasets. Currently, we are working on extending these algorithms to metagenome datasets and to 3rd generation single-molecule sequencing data. In particular, longer but more error prone 3rd generation single-molecule sequencing data might require different graph analytical abstractions (string graphs instead de Bruijn graphs). Metagenomic datasets require significant modifications to the data processing pipelines, performing multiple sweeps over the data in an iterative streaming manner to peel out genomes of different species.

MANTISSA contributed to the development of HipMer (https://sourceforge.net/projects/hipmer/), an end-to-end high performance de novo assembler designed to scale to massive concurrencies. We have shown that HipMer can be applied even to genomes such as bread wheat that are larger and more complex than the human genome. This opens up opportunities both for routine genome assembly of plants and animals of agricultural value. By associating key traits (e.g., yield, drought tolerance) to genetic variation, crops and livestock can be improved by genome-assisted breeding. High throughput large-scale genome assembly also enables a broader sampling of biodiversity, facilitating projects like the “Genomes10K” program that is focused on sequencing a representative sampling of 10,000 vertebrate species, the i5K Consortium focusing on sequencing 5000 insects and arthropods, and other programs to sequence 3000 strains of rice or cataloging 1000 fungi.

-
Evangelos Georganas, Aydın Buluç, Jarrod Chapman, Steven Hofmeyr, Chaitanya Aluru, Rob Egan, Leonid Oliker, Daniel Rokhsar and Katherine Yelick, "HipMer: An Extreme-Scale De Novo Genome Assembler". 27th ACM/IEEE International Conference on High-Performance Computing, Networking, Storage and Analysis (SC 2015), Austin, TX, USA, November 2015.
-
Evangelos Georganas, Aydın Buluç, Jarrod Chapman, Leonid Oliker, Daniel Rokhsar and Katherine Yelick, "merAligner: A Fully Parallel Sequence Aligner". 29th IEEE International Parallel & Distributed Processing Symposium (IPDPS 2015), Hyderabad, INDIA, May 2015.
-
Jarrod A Chapman, Martin Mascher, Aydın Buluç, Kerrie Barry, Evangelos Georganas, Adam Session, Veronika Strnadova, Jerry Jenkins, Sunish Sehgal, Leonid Oliker, Jeremy Schmutz, Katherine A Yelick, Uwe Scholz, Robbie Waugh, Jesse A Poland, Gary J Muehlbauer, Nils Stein and Daniel S Rokhsar "A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome" . Genome Biology 2015, 16:26 .
-
Evangelos Georganas, Aydın Buluç, Jarrod Chapman, Leonid Oliker, Daniel Rokhsar and Katherine Yelick, "Parallel De Bruijn Graph Construction and Traversal for De Novo Genome Assembly". 26th ACM/IEEE International Conference on High-Performance Computing, Networking, Storage and Analysis (SC 2014), New Orleans, LA, USA, November 2014.