




Machine Learning Algorithms Enhance Predictive Modeling of 2D Materials
March 2, 2017
By Kathy Kincadez
Contact: cscomms@lbl.gov
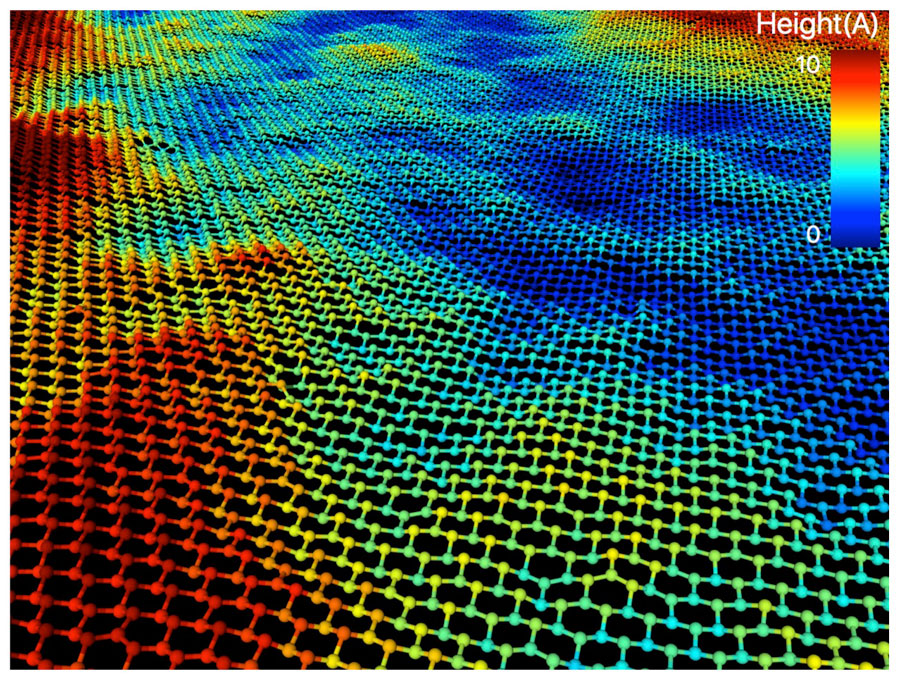
Stanene is softer and consequently much more rippled than its cousins graphene and silicene. Image: Mathew Cherukara, Badri Narayanan and Subramanian Sankaranarayanan, Argonne National Laboratory.
Researchers from Argonne National Laboratory, using supercomputers at Berkeley Lab’s National Energy Research Scientific Computing Center (NERSC), are employing machine learning algorithms to accurately predict the physical, chemical and mechanical properties of nanomaterials, reducing the time it takes to yield such predictions from years to months—in some cases even weeks. This approach could help accelerate the discovery and development of new materials.
Using a modeling framework built around a molecular dynamics code (LAMMPS), the research team ran a series of simulations to study the structure and temperature-dependent thermal conductivity of stanene, a 2D material made up of a one-atom-thick sheet of tin. This work, which involved a set of parameters known as the “many-body interatomic potential” or “force field,” yielded the first atomic-level computer model that accurately predicts stanene’s structural, elastic and thermal properties. The findings were published in The Journal of Physical Chemistry Letters.
In addition, by applying machine learning algorithms, the research team was able to achieve these characterizations much faster than ever before. Traditional atomic-scale materials models have taken years to develop, and researchers have had to rely largely on their own intuition to identify the parameters on which a model would be built, noted Subramanian Sankaranarayanan, a computational scientist at Argonne and team leader on The Journal of Physical Chemistry Letters paper. But by using machine learning, the Argonne researchers were able to reduce the need for human input while shortening the time to craft an accurate model down to a few months.
“Characterizing a force field like this takes a really long time to do by hand—a matter of years—so machine learning is a way of automating this process, speeding it up, so that we can explore more of the parameter space than a human can do and get a faster, more accurate fit,” said Mathew Cherukara, an Argonne postdoctoral researcher and lead author on the study.
The researchers used supervised machine learning algorithms such as Genetic Algorithms combined with Simplex for optimization—algorithms that have been around for 15-20 years and are commonly used in industries such as banking and social media. “We are now applying this to materials, and that has not been done before,” Cherukara added. “And using these machine-learnt force fields, we can do more than these materials models were originally designed to do.”
Framework Not Material-Dependent
Unlike previous atomic-scale materials models, the machine learning approach can capture bond formation and breaking events accurately, yielding more reliable predictions of material properties such as thermal conductivity) and enabling researchers to capture chemical reactions more accurately and better understand how specific materials can be synthesized.
“We input data obtained from experimental or expensive theory-based calculations and then ask the machine, ‘Can you give me a model that describes all of these properties?’” said Badri Narayanan, an Argonne postdoctoral researcher and another lead author on the study.
In addition, because the process is not material-dependent, researchers can look at many different classes of materials and apply machine learning to various other elements and their combinations.
“What surprised us is that we can get more out of these models than we thought possible,” Cherukara said. “We can use, for example, the same model framework that we used for stanene to describe water—two very different materials. The underlying analytical model is the same; the difference is the numbers that go in and the parameterization of the model.”
Another surprise is how well the machine learning framework learns outside of what it is originally trained against.
“We found that if you take this model and start working on data that was not used for training, it actually does very well outside of where it was originally trained against,” Sankaranarayanan said. “The framework is so robust that it allows us to develop models well beyond what they have been trained against. And that is what you want it to be: very good in new scenarios that were not in the original training. We generally expect this model to have limited predictive power, but when you start using it in different scenarios, the predictions actually come out very good.”
The framework is already being used by other researchers at Argonne, and Sankaranarayanan and his collaborators envision it eventually being something even non-experts can use to explore materials properties.
“Right now it takes a certain amount of expertise to use it, but our goal is that pretty much anyone can use it,” Narayanan said. “The main framework already exists and is very useful for materials research; it just needs to be made a little more user friendly.”
Part of the work was performed at the Center for Nanoscale Materials (CNM) at DOE’s Argonne National Laboratory. Both CNM and NERSC are DOE Office of Science User Facilities.
This article was adapted from materials provided by Argonne National Laboratory.
About NERSC and Berkeley Lab
The National Energy Research Scientific Computing Center (NERSC) is a U.S. Department of Energy Office of Science User Facility that serves as the primary high performance computing center for scientific research sponsored by the Office of Science. Located at Lawrence Berkeley National Laboratory, NERSC serves almost 10,000 scientists at national laboratories and universities researching a wide range of problems in climate, fusion energy, materials science, physics, chemistry, computational biology, and other disciplines. Berkeley Lab is a DOE national laboratory located in Berkeley, California. It conducts unclassified scientific research and is managed by the University of California for the U.S. Department of Energy. »Learn more about computing sciences at Berkeley Lab.